

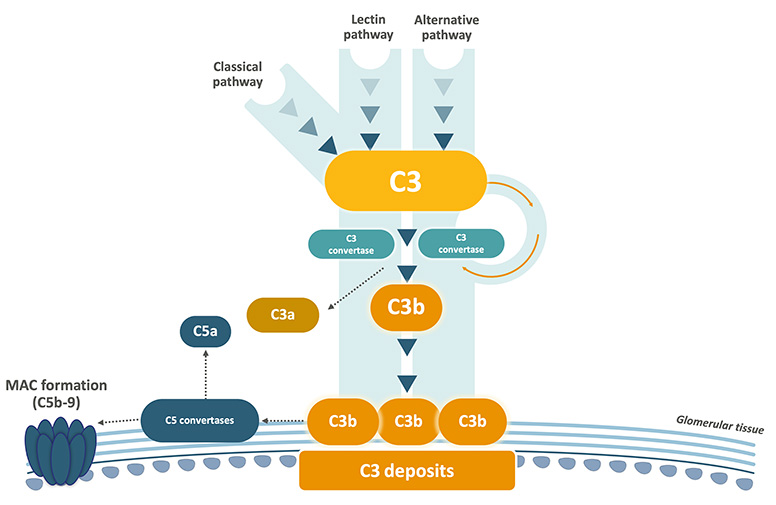
Both C3G and primary IC-MPGN are characterised by C3 dysregulation resulting in overactivation of C3, leading to the accumulation of C3 breakdown products in the glomeruli. This triggers inflammation and structural damage, resulting in progressive kidney injury.2,5
The disease begins with an initial trigger, such as an infection or an autoimmune condition, initiating C3 dysregulation. C3 deposits may accumulate in the kidney before clinical symptoms appear, as shown in animal models.4
Genetic and Immunological Factors
While the exact cause of complement dysregulation remains unknown in many patients, contributing factors have been identified:1,3
- Genetic mutations in complement-related genes (e.g., C3, CFH, CFI, CFB, CFHR) are found in 10–20% of cases. Familial cases are very rare, indicating that C3G is not a monogenic disease for most patients.1,3
- Complement nephritic factors (CNeFs), are present in up to 50% of patients. CNeFs are auto-antibodies which stabilise complement convertases, prolonging their half-life, resulting in excessive C3b deposition.1,4,6
- C3NeFs (stabilise the C3 convertase) are the most common, but patients may also present with C5NeFs and less frequently with C4NeFs. In addition, autoantibodies against complement components such as Factor H and Factor B, are occasionally detected.3
The accumulation of C3 deposits seen in C3G and primary IC-MPGN is associated with the recruitment of inflammatory cells to the glomerular compartments, disrupting membrane function and causing glomerulonephritis (inflammation of the glomeruli).1,2
Glomerulonephritis causes ongoing glomerular injury and rupture of the glomerular basement membrane, detectable by leakage of serum proteins (proteinuria), erythrocytes (haematuria), and leukocytes into the urine.2
Glomerulonephritis may also present with nephrotic syndrome (massive proteinuria and oedema), if podocyte damage is significant.2,3
The consequences of persistent glomerulonephritis caused by C3 dysregulation can include scarring, tubular atrophy and glomerulosclerosis, resulting in chronic kidney disease and irreversible kidney failure.1,2,4
Although both diseases involve C3 dysregulation, they are different disease entities which differ in their immunopathology.7,8
Primary IC-MPGN is distinguished by the substantial presence of Ig deposits, indicating a predominant activation of the complement classical pathway.9
C3G and primary IC-MPGN can appear identical with light microscopy, but immunofluorescence tells us they are not the same disease.1,2
- C3G is defined by dominant C3 staining (≥2 orders of magnitude greater than any other immune reactant) with minimal or no immunoglobulin deposition.7-9
- Primary IC-MPGN features both C3 and immune complex (Ig) deposits.7-9
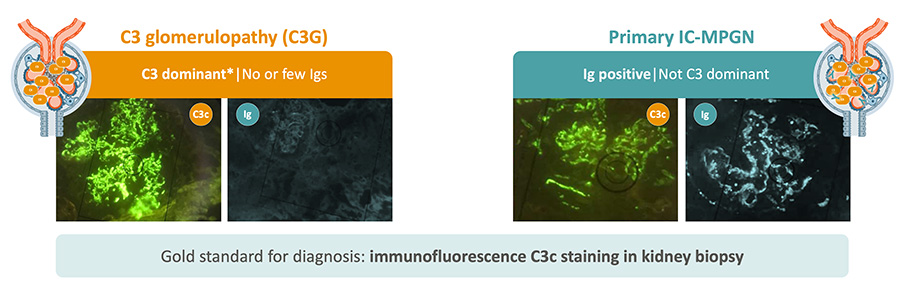
*C3 dominant: C3 is ≥2 orders of magnitude stronger than for any other common immune reactant.
C3, complement 3; C3G, C3 glomerulopathy; CFB, complement factor B; CFH, complement factor H; CFHR, complement factor H-related protein; CFI, complement factor I; CNeF, complement nephritic factor; IC-MPGN, immune complex-mediated membranoproliferative glomerulonephritis.
References:
- Meuleman MS, et al. Semin Immunol 2022;60:101634.
- Smith RJH, et al. Nat Rev Nephrol 2019;15:129–43.
- Xiao X, et al. Mol Immunol 2016;77:89–96.
- Fakhouri F, et al. Kidney Int 2020;98:1135–48.
- Schena FP, et al. Int J Mol Sci 2020;21:525.
- Michels MAHM, et al. Front Immunol 2022;13:1036136.
- Fervenza FC, Sethi S, Glassock RJ. Nephrol Dial Transplant 2012;27(12):4288–94.
- Cook HT, Pickering MC. Nat Rev Nephrol 2015;11(1):14–22.
- Noris M & Remuzzi G. Nephrol Dial Transplant 2024;39:202-14
- Mastrangelo A, et al. Front Pediatr 2020;8:205.